Institute of Molecular Genetics of the Czech Academy of Sciences
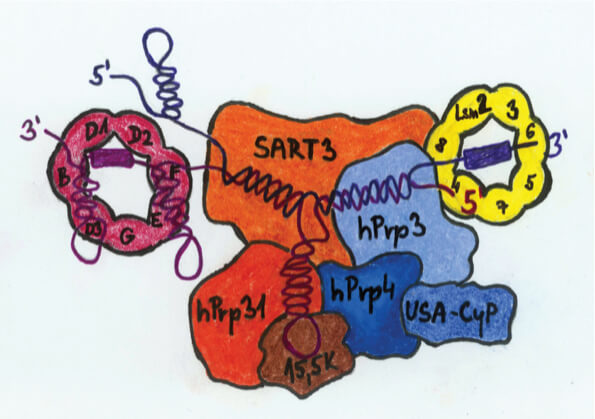
Our DNA contains the information for the synthesis of all our proteins. However, this information in human DNA is fragmented and our genes contain long, seemingly “useless” sequences that need to be removed in a process called RNA splicing. The useless RNA sequences are removed by a large, sophisticated and dynamic molecular machine called the spliceosome. The spliceosome is one of the most complex particles in our cells, composed of several non-coding RNAs and ~150 accessory proteins. Our long-term goal is to determine how the spliceosome assembles at the right time and place inside the cell. We are investigating how nuclear architecture contributes to the correct spliceosome formation, and studying the molecular principles of the control mechanism that distinguishes correctly assembled spliceosome particles from the defective ones. Finally, we seek to determine why mutations in several ubiquitously expressed spliceosomal components cause retinitis pigmentosa, a human genetic disease characterized by photoreceptor cell degeneration.